Written by: Matt Baran-Mickle
If you haven’t already, be sure to check out Part 1.
The Intestinal Barrier: Where the Rubber Meets the Road
The mucosal immune system[1] is the largest interface between our internal and external environments – the small intestine alone has an approximate surface area of 400 m2 – and is responsible for the elimination of a diversity of pathogens while maintaining tolerance to large populations of resident bacteria and the continuous flow of food-derived particles that pass through the digestive system every day. While this system includes everything from the epithelia of the urogenital tract, the respiratory tract, and the mammary glands to that that of the digestive tract, the mucosal immune system of the intestines is of primary importance when considering the impact of nutrition on immunity.
The intestinal mucosa is home to a wide variety of leukocytes, including specialized populations of lymphocytes that are site-specific. Together, these cells coordinate a fine balance between tolerance and reactivity, and in healthy mucosa inflammation is largely absent, demonstrating the effectiveness of local homeostatic control given the large numbers and volume of potential pathogens passing through the intestines every day, and the presence of 1012 bacterial cells/mL in the colon contents.
Celiac disease, barrier disruption, and inflammation
Disruption of this sensitive intestinal barrier is associated with a number of diseases, most notable Celiac disease (CD) and the inflammatory bowel diseases (IBDs: Crohn’s disease and ulcerative colitis). CD is an increasingly prevalent autoimmune/allergic disease that is triggered in genetically susceptible individuals by the ingestion of the gliadin fraction of gluten, a protein found in wheat, rye, oats, and barely, and similar proteins found in other cereal grains. The prevalence of CD in the US was recently established as 1 in 133, with a higher incidence in close relatives of CD patients.
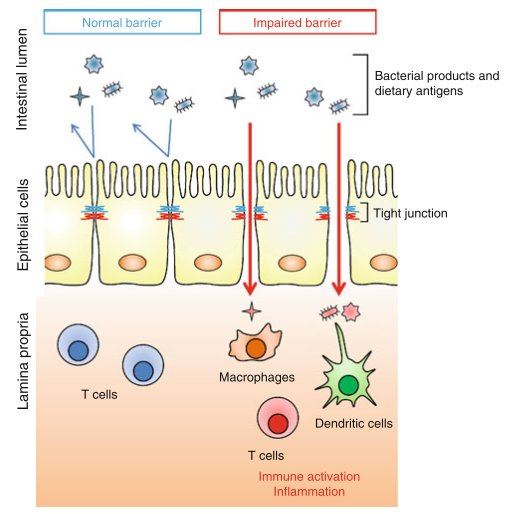
(image: entry of luminal antigens with disruption of tight junctions; Suzuki, 2013).
CD shares comorbidity with a number of other autoimmune diseases, most notably type 1 diabetes (an autoimmune disease of the pancreas), and provides an example of the potential impact of a bioactive food protein on the homeostasis of the intestinal mucosa. In CD, digestion of gliadin into peptide fragments allows them to bind to receptors on intestinal epithelial cells, beginning an intracellular signaling cascade that loosens the structural connections between epithelial cells, called tight junctions (TJs). TJs normally function to prevent the passage of the luminal contents into the connective tissue lying underneath the epithelium, and the disruption of TJ integrity can allow them access to leukocytes that are normally isolated from the luminal contents
In CD, gliadin fragments are taken up by cells of the innate immune system and presented to T cells, generating an adaptive response to gliadin and the production of inflammatory cytokines by innate cells. This is mediated by specialized molecules of the innate immune system that “present” peptide fragments to T cells, dubbed MHC molecules after the gene that encodes them, the major histocompatibility complex. When gliadin makes its way into the epithelium, it is modified by the enzyme tissue transglutaminase (tTG), converting the amino acid glutamine into glutamate and allowing it to bind more effectively to MHC molecules.
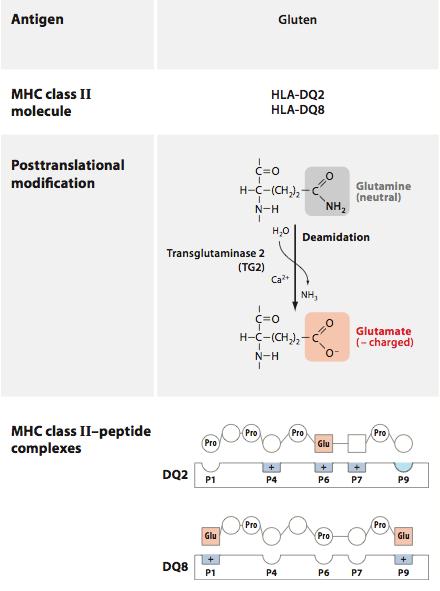
(image: deamidation of gliadin – DQ2 and DQ8 are risk alleles for CD; Abadie, Sollid, Barreiro, & Jabri, 2011).
Certain alleles of these molecules constitute the genetic risk factors for celiac, as their high affinity for gliadin fragments allows for enhanced recognition by T cells. Once activated, these T cells promote inflammation that recruits additional leukocytes to the small intestine, and promotes the production of antibodies that target gliadin and self-proteins in the local environment. T cells that are activated in this context can target epithelial cells for destruction, leading to the intestinal damage found in biopsies of CD patients.
Inflammatory cytokines can themselves alter TJ function, worsening the barrier dysfunction initiated by gliadin signaling, and generating a feed-forward loop of TJ disruption and further inflammation. The mucosae possess a variety of self-reactive lymphocytes, and these may escape into the circulation, allowing them access to tissues that they can target for destruction. Under physiological conditions this is prevented by Treg cells and other tolerance-promoting mechanisms, but the presence of inflammation can provide an activating signal that is normally absent.
This mechanism of activation, and the potential to generate an autoimmune attack on tissues beyond the intestines, is hypothetical at this point, but the presence of barrier disruption in a number of autoimmune diseases provides support for the concept.
Gut flora and regulation of the mucosal immune system
The existence of large numbers of bacteria in our large intestines, and, to a lesser extent, our small intestines, has long been recognized, but the impact of the microbiota (colloquially, gut flora) on human health has only recently been targeted as a major area of research. The development of new culture-independent techniques has allowed researchers to rapidly quantify the composition of the microbiota and compare profiles between diseased and healthy populations, as well as track the development of the microbiota with age. Investigations into the impact of the microbiota on immune function complement this work, and contribute to a developing, though still nascent, picture of the functional relationship between bacteria and their hosts in homeostasis and disease. Research has identified potential roles for microbiota in a growing number of conditions, including inflammatory bowel disease, asthma and allergic diseases, rheumatoid arthritis, mouse models of multiple sclerosis, and type 1 diabetes.
The gut flora, under physiological conditions, are symbiotic and promote the proper development of the mucosal immune system and its proper function once established. Cells of the innate immune system regularly sample luminal contents in the intestines, and the recognition of bacteria by these cells provides activating signals that generate a baseline level of inflammation (physiological inflammation) that is crucial in maintaining a proper mucosal barrier. The importance of these interactions are evident in germ-free mice raised without bacterial exposure that demonstrate altered immune responses and reduced numbers of Treg cells in the large intestine.
Furthermore, bacteria produce a number of metabolites that promote mucosal homeostasis, including short-chain fatty acids that are the primary fuel source of colonic epithelial cells. The gut flora rely on nutrients that we ingest in order to survive, and changes in diet are accompanied by changes in our bacterial communities. Bacteria possess a number of enzymes that can metabolize plant polysaccharides that we are incapable of digesting, and a diet low in plant material fails to support these potentially important bacterial species. A key role of commensal bacteria is to prevent colonization by pathogenic bacteria, and dysbiosis generated by a low-polysaccharide diet may allow the infiltration of harmful bacterial species, the promotion of inflammation, and the subsequent activation of self-reactive lymphocytes, as discussed above.
The concept of dysbiosis – a pathological imbalance of intestinal bacterial populations – as a possible mechanism of disease is gaining traction, and evidence that dysbiosis can impact TJ integrity is a key observation. While continual stimulation by the gut flora is necessary for proper immune function, inflammation beyond physiological levels may hamper the negative feedback mechanisms that are in place to prevent penetration of bacteria into the epithelium. Excessive exposure to luminal bacteria may, once again, generate a feed-forward inflammatory loop that serves to further damage the integrity of the mucosal barrier.
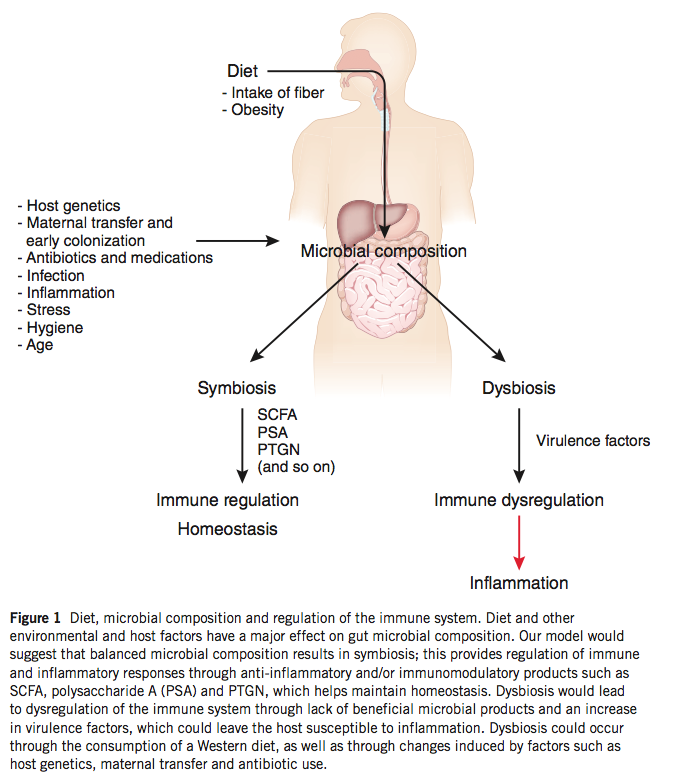
(image: impacts of environmental factors on the gut flora; Maslowski & Mackay, 2011)
An important detail to keep in mind in this discussion is the ability of systemic inflammation from other sources to alter TJ integrity and damage mucosal barrier integrity and to change the composition of bacterial communities independently. Dysbiosis may be a driver of disease, but can also be a consequence of inflammation and its effects on the mucosa. While it is tempting to pin the source of inflammation (and subsequent activation of self-reactive cells) on bioactive plant proteins like gliadin and a disrupted microbial community, other sources of inflammation may precede changes in the mucosal immune system. One such source of inflammation is obesity, the prevalence of which in industrialized society makes it a likely contributor to the epidemic of autoimmune disease we see today.
[1] The “mucosal epithelia” refers to those epithelial barriers lining the body’s internal cavities (e.g. the digestive tract) that secrete a protective layer of mucus. The mucosal immune system is a distinct compartment of the immune system associated with the mucosal epithelia.
Image credits:
Abadie, V., Sollid, L. M., Barreiro, L. B., & Jabri, B. (2011). Integration of genetic and immunological insights into a model of celiac disease pathogenesis. Annual review of immunology, 29, 493–525. doi:10.1146/annurev-immunol-040210-092915
Maslowski, K. M., & Mackay, C. R. (2011). Diet, gut microbiota and immune responses. Nature immunology, 12(1), 5–9. doi:10.1038/ni0111-5
Suzuki, T. (2013). Regulation of intestinal epithelial permeability by tight junctions. Cellular and molecular life sciences?: CMLS, 70(4), 631–59. doi:10.1007/s00018-012-1070-x
Matt Baran-Mickle graduated from Hampshire College in 2013 with a BA in Human Physiology and Immunology, completing a senior thesis investigating the connections between nutrition and autoimmune disease from an immunological perspective. He completed pre-med requirements during his undergraduate work, and hopes to pursue a degree in Naturopathic medicine beginning in 2015. In the meantime, he eats, trains, and works in Boulder, CO.

Quality info, Matt! Keep it coming.
So where does this leave people with IBD who want to do keto / zero-carb?
Where does that leave all the other paleo carnivores out there? Will they have to start eating uncooked potato starch (like Richard Nikoley blogged about)?
Are they forever condemned to a life of thin, demure mucuous?
A concise yet thorough set of articles. Very well presented, you’ve done your research, thank you for sharing it with us.
Although there is so much written about avoiding FODMAPS, it is clear to me that our cohabitants need them, I am glad to see you point that out.
I do have some related queries, if you are so inclined to answer…
1) when you eat prebiotics/FODMAPS( assuming you are not sensitive) which your good bacteria need to thrive – don’t you simultaneously feed the pathogenic bacteria as well, and you may be promoting dysbiosis
2) With the ingestion of probiotics, why do the good bacteria not “set up shop”in, ie colonize, your intestine as other bacteria do (such as the pathogenic ones)? Why are they transients and therefore you require daily intake of probiotic to maintain gut health?
I may be asking questions for which definitive answers are not known, I just want to know if I am missing something obvious
You are correct on point 1 and this is one of the challenges of getting this “right.” Some folks do GREAT with fodmaps, for others it’s a trainwreck. As to point 2, it seems like constant exposure to new bacteria is the ancestral norm…I suspect it’s both normal and healthy for the gut flora to be labile.
While it’s recognized that most probiotics exert their therapeutic effects while in transit, products like Prescript Assist (a probiotic recommended by Chris Kresser) made up of soil-based organisms may actually colonize our intestines instead of passing right through. As far as the “why,” I don’t have a good answer, all we have right now are the observations that they don’t. In my reading of the literature I haven’t come across a good explanation of your first point (as Robb noted), but there may be slightly different substrate (food) preferences for pathogenic bacteria vs. beneficial bacteria, and the pathogenic bacteria may be less common, making it easier to use a blanket approach to support the good guys.